1. Introduction
entinogenesis imperfecta (DI) is an autosomal dominant genetic condition characterised by abnormal dentin structure affecting either the primary or both the primary and secondary dentitions. Incidence of DI is 1 in 6000 to1 in 80001.Clinically teeth are opalescent with the color ranging from bluish-gray to brown to yellowish and exhibit pronounced attrition of incisal and occlusal edges. Radiographically, crowns are bulbous with cervical constrictions and the roots are short. The pulp chambers and root canals are usually obliterated due to dentin over production. Synonyms are-Hereditary opalescent dentine, Capdepont teeth.
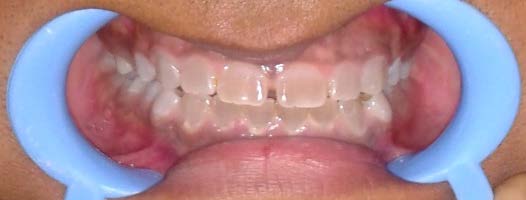
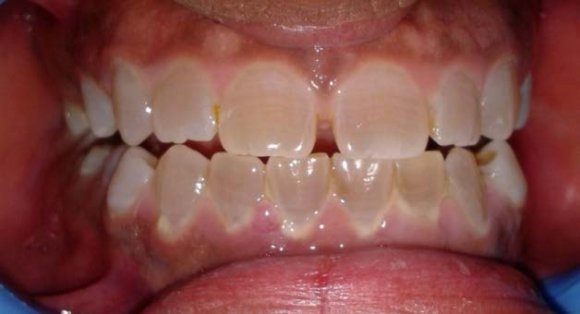
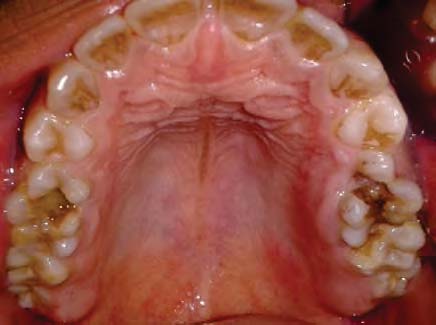
Here we report a case of 21 year old male presenting with DI type II with clinical and radiographic features.
2. II.
3. Case Report
4. Discussion
Dentinogenesis imperfecta (DI) is one of the most common genetic disorders affecting the structure of dentin. It was recognized first time by Barret in1882 .In 1887 the condition was first described in a case involving a completely normal boy with dark staining on the teeth. 2 The term 'Dentinogenesis imperfecta' was coined by Robert and Schour in 1939. 3 Witkop named the types Dentingenesis imperfecta, Hereditary opalascent dentin, and Brandywine isolate 4 . S Sheild's et al classified Dentinogenesis imperfecta based on phenotypic variability into-TypeI, Type II and Type III. Type I-occurs with Osteogenesis imperfecta. TypeII-DI not associated with Osteogenesisimperfecta; also known as hereditary opalescent dentin. Type III-DI of the "Brandywine type" 5 . Brandywine type (Dentinogenesis imperfecta 2) was found in the Brandywine triracial isolate in Southern Maryland. Multiple pulp exposures and "shell teeth" (due to large pulp chambers and thin dentinal walls) are two characteristics used to distinguish DI type III fromDI typeII. Genetic research has confirmed that Osteogenesis imperfecta with opalescent teeth clearly is a separate disease from Dentinogenesis imperfecta. Osteopontin,a bone glycoprotein is also expressed in dentin. However, there is no association between a type of polymorphism at the osteopontin locus and dentinogenesis imperfecta. Hence the following revised classification is proposed.
Dentinogenesis imperfecta1: Dentinogenesis imperfecta without osteogenesis imperfecta: corresponds to dentinogenesis imperfecta type II of Shields classification.
Dentinogenesis imperfecta 2: Brandywine type dentinogenesis imperfecta: This corresponds to dentinogenesis imperfcta type III of Shields classification.
There is no substitute in the present classification for the category designated as DI type I of the Shields classification.
Clinically both the dentitions exhibit an unusual translucent, opalascent appearance with colour ranging from yellow-brown to grey. The entire crown appears discoloured because of the abnormal underlying dentin. Excessive constriction is present at the cementoenamel junction, giving the crowns a tulip or bell shape. Enamel is structurally and chemically normal but fractures easily which leads to rapid wear. The enamel fracturing is believed to be due to the poor support provided by the abnormal dentin and possibly in part to the absence of the scalloping normally seen between dentin and enamel that is believed to help mechanically lock the two hard tissues together 6 . The micohardness of the dentin closely approximates that of cementum, which also results in rapid attrition 8 .Rapid wearing and absence of interdental contacts make the teeth less susceptible to caries. Dental tissues in DI will have low hardness, elasticity and stiffness leading to a phenomena of micromovement resulting in failure of restorations. 7 Radiographically opacification of dental pulps occur in both the types I and II because of continued deposition of abnormal dentin. Microscopically, dentin in dentinogenesis imperfecta contains fewer, but larger and irregular dentinal tubules. Pulp is nearly completely replaced over time by the irregular dentin. Enamel appears normal, but the DEJ is smooth instead of scalloped 6 .
Dentin has two proteins in its composition: DSPP (dentinphospho protein) and dentin sialoprotein (DPS). DSPP is expressed in a number of tissues including bone, kidney, salivary gland and lung but its expression in dentine is hundreds of times higher than in other tissues Disturbances in the secretion of these proteins, and thus in the proper shape and placement of dental matrix crystals of apatite, manifested clinically as dentinogenesis imperfecta 9 .The genes responsible for producing both DPP and DSP are located at 4q12-21. Type I and Type III of DGI appear to result from mutations in the gene encoding DSPP suggesting that these conditions are allelic. 10 associated with dentinogenesis imperfecta are Osteogenesis imperfecta, Ehlers Danlos syndrome, Goldblatt syndrome, Schimkeimmunoosseousdysplasia, Brachio-skeletogenital syndrome, Osteodys plastic primordial short stature with severe microdontia, opalescent teeth, and rootless molars 11 Dentinogenesis imperfecta should be differentiated both clinically and radiographically from Amelogenesis imperfect (AI), Regional odonto dysplasia, Dentin dysplasia (DD), Tetracycline staining, Irradiation to jaws or chemotherapy during root development, Congenital erythropoietic porphyria and Dental Fluorosis. Amelogenesis imperfecta like DI, is also a hereditary disorder. In AI teeth are usually sensitive and on radiographs enamel is less radio-dense and thinner than dentin. Pulp chamber and Root canals are usually not sclerosed.
Regional odontodysplasia is a localised anomaly restricted to a single tooth or a group of contiguous teeth while in dentinogenesis imperfecta all the teeth are involved. In Regional odontodysplasia the involved teeth either exhibit delayed eruption or do not erupt at all. Pulp chamber is very large giving a pale hazy image to the affected teeth, which is termed as ghost teeth.
Dentin dysplasia -Both DI and DD can produce crowns with altered colour and occluded pulp chambers. The finding of a 'thistle tube's haped pulp chamber in single rooted tooth strengthens the possibility of dentin dysplasia. The crowns in dentin dysplasia are usually of normal shape, size and pe e t
The type II ec ec a of si 9 .T at at e e a i im r Syndromes proportion while in dentinogenesis imperfecta teeth have bulbous shaped crowns with a constriction in the cervical region. If the roots are short and narrow, the condition is likely to be dentinogenesis imperfecta. On the other hand, normal appearing roots are present in dentin dysplasia type II or practically no roots at all in dentin dysplasia typeI12 `Congenital erythropoietic porphyria-It is a rare condition resulting from an inborn error of porphyrin metabolism. Abnormally high levels of porphyrin pigments are incorporated into teeth during their formation The entire primary and secondary dentitions are pink or reddish brown. Under ultraviolet light, the teeth fluoresce red13.The teeth discolouration is usually found at the necks of teeth and the enamel hypoplasias are usually located in coronal third of the teeth and no pulp sclerosis is seen while in DI pulp sclerosis is present.
Tetracycline staining-The erupting affected teeth have a bright yellow band-like appearance that fluoresces under ultraviolet light. On exposure to sunlight, the colour gradually changes to grey or redbrown. Radiographically there is no pulp sclerosis in tetracycline staining while in Dentinogenesis imperfecta pulp sclerosis is present.
Dental Flourosis-Ingestion of drinking water containing fluoride at levels greater then 1 ppm during the time crowns are being formed may result in enamel hypoplasia or hypocalcification or fluorosis. Mild to moderate fluorosis ranges clinically from white enamel spots to mottled brown and white discolorations. Severe fluorosis appears as pitted, irregular and discoloured enamel. No pitting is seen in dentinogenesis imperfecta and also crown gives opalescent appearance.Pulp obliteration is seen in dentinogenesis imperfecta which is absent in dental fluorosis.
5. IV.
6. Conclusion
Dentinogenesis imperfecta causes esthetic as well as functional problems to the patients.Where diagnosis occurs early in the life of the patient,good aesthetics and function can be obtained thereby minimising nutritional deficits and psychosocial distress.To give a diagnosis of dentinogenesis imperfecta syndromes related to it and other causes of teeth discolouration should be taken into consideration.