1. Introduction
he Bohring-Opitz syndrome (BOPS) was first described in 1999 by Bohring et al (1). It is an extremely rare genetic condition, of unknown prevalence, which is caused by de novo or nonsense mutations in the ASXL1 gene. To date 46 people with BOPS have been described, of whom only 20 have a confirmed molecular diagnosis. The BOPS diagnosis is established by clinical suspicion and / or identification of a constitutional heterozygous pathogenic variant in the ASXL1 gene (2). This article shows the first case in Latin America of BOPS confirmed by molecular diagnosis.
2. II.
3. Case Report
We present an 8-year-old male patient, born to term at 38 weeks, presented intrauterine growth restriction with birth weight of 2200 grams (P 0.4, Z -3.1) and a birth lenght of 48 centimeters (P 17, Z 0.95). He was hospitalized for 40 days in the neonatal intensive care unit (NICU) where he received mechanical ventilation for the first six days. During his hospital stay in NICU an echocardiogram was performed and a moderate mitral regurgitation, a moderate pulmonary hypertension and an ostium secundum interatrial communication were identified. No evidence of hemodynamic repercussion was found. At one year of age a ventricular dilatation are evidenced in brain MRI. Developmental delay was documented. Physical exam showed discrete synophris, ocular hypertelorism, myopia, bilateral sensorineural hearing loss, cleft palate, velo-palatal incoordination, cryptorchidism, left thoracolumbar scoliosis, pre-axial polydactyly in the hands and hypotonia. Current athropometry was: weight 25 kg (P21, Z -0.8) and height 120 cm (P2, Z -2.1), IMC 17.4 (P75, Z 0.67)
In the first year of life, severe uptakeswallowing disorder managed with gastrostomy is documented; later, in the preschool age he presented multiple hospitalizations due to infectious diseases, pneumonias and recurrent otitis, because of this, inmmunodeficiency was ruled out. He developed symptomatic refractory epilepsy that was treated with carbamazepine and topiramate. Finally, Bohring-Opitz syndrome was suspected [25], a complete sequencing of the ASLX1 gene was performed, identifying the heterozygous variant C.2893C>T (p.Arg965*) that confirmed the diagnosis of BOPS.
4. III.
5. Discussion
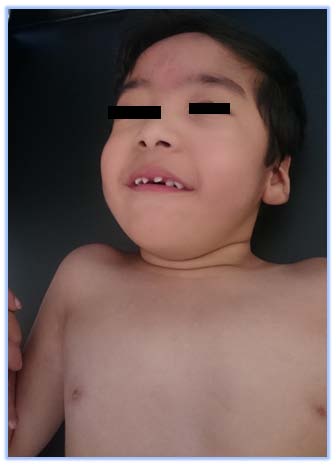
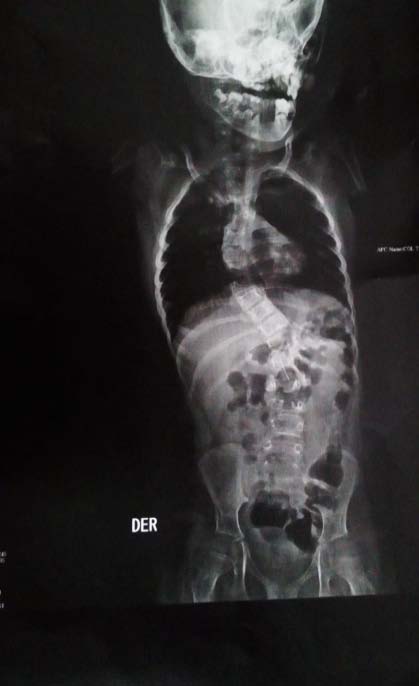
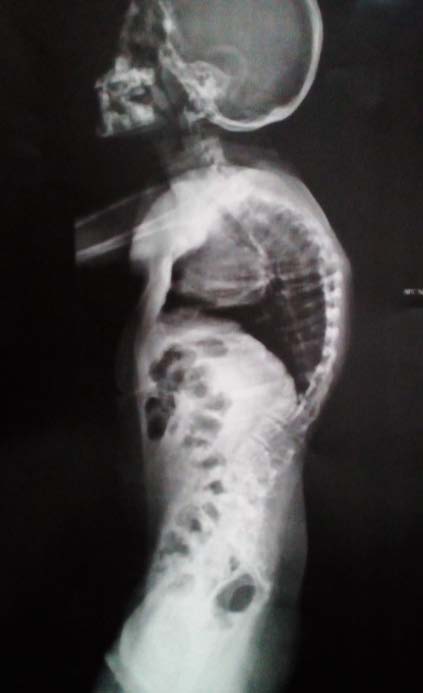
The Bohring-Opitz Syndrome (BOS, OMIM 605039) is caused by de novo or nonsense mutations in the ASXL1 gene (OMIM 612990) (3), that accounts for about 50% of the cases that meet the clinical criteria. The gene locus is located on chromosome 20q11.21, contains 13 exons and codes for the sex-combs-like 1 protein, which is a polypeptide of 1543 amino acids (2,4). The sex-combs-like 1 protein is involved in the remodeling of chromatin in localized areas and helps to activate and silence the transcription of different genes involved in the regulation of the expression of HOX genes, involved in embryonic development for determination of the basic structure and orientation of the embryo (5), which proposes a mechanism of loss of function, that is, haploinsufficiency, as the fundamental cause of the BOPS clinical picture. (3) To date, genotype-phenotype correlations have not been reported because of the few number of individuals in whom pathogenic variants in the ASXL1 gene have been identified (2). The described phenotype of the BOPS includes distinctive facial and postural features, delayed neurodevelopment, failure of thrive and other associated clinical conditions of variable presentation. The facial features are distinctive and include pronounced microcephaly in the early years of childhood, trigonocephaly that is generated from the prominence of the metopic ridge and a narrowing at the bitemporal level (6), hypotonic facies, "nevus flammeus", telecanto, hypoplastic supraorbital crests, upward slanting palpebral fissures, depressed nasal bridge, anterverted nostrils, hairy and arched eyebrows, posteriorly rotated ears, hypertrichosis, micrognathia and narrow and high palate (see figure 1) (7). Facial manifestations tend to become less noticeable with age. ( 8) At the ocular level, ptosis, strabismus that does not resolve, and glaucoma due to pressure increase at the level of the anterior chamber of the eye are evident. (7) Similarly, alterations of the retina and the optic nerve, which include colobomas, optic nerve atrophy, atrophy of the retina and / or abnormal pigmentation that explain visual impairment (2, 9) are common. Patients diagnosed with BOPS who have a mutation identified in the ASXL1 gene have a higher incidence of myopia (87% versus 40%) and hypertrichosis (89% versus 17%), compared to those without mutations. (5) A typical BOPS posture that is commonly identified in early childhood and becomes less apparent with age has been described: it presents with shoulders directed towards the midline, extremities flexed distally (elbows, wrists and fingers) forming a fixed contracture position (see figure 2) (6). Despite the typical posture, no studies have been found that relate it to CNS alterations, muscle tone or joint dislocations (1). In addition, alterations in muscle tone ranging from flaccidity to hypotonia of the upper and lower extremities leading to alterations in the curvatures of the spine are described (Figure 3) (6). Similarly, congenital contractures, dislocations and pectus excavatum are observed (2); the congenital dislocation of the hip and the radial head are reported in up to 33% of the cases, the first was evidenced in the present patient (Figure 4). In the central nervous system, ventriculomegaly, delayed myelination, Dandy-Walker malformation and generalized atrophy with abnormalities in neuronal migration that favors the development of seizures have been identified (4). These findings may explain the variable intellectual disability, the deficit of language and the difficulties for independent bipedestation. Very few achieve an independent walking.(6) It is described that children with BOPS can recognize caregivers and have a social and interactive nature, so they are seen with a happy and pleasant attitude.
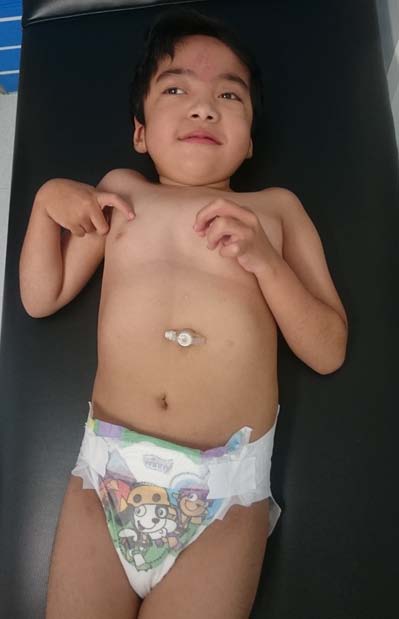
Patients with BOPS have swallowing difficulties since early childhood, functional constipation, gastroesophageal reflux (GER), cyclical vomiting and poor suction; which are related to poor weight gain and are thought to be have a neurogenic origin (10). Gastrointestinal disorders are observed from the first months of life and tend to improve with age. Nevertheless, 53% of the patients require parenteral nutrition or gastrostomy as a definitive form of nutrition (Figure 2), and have a risk of aspiration and dehydration.
Half of patients may have minor cardiac anomalies, transient bradycardia and apnea (2). Similarly, septal and cardiac hypertrophy, persistent ductus arteriosus and valvular anomalies, pulmonary stenosis being the most representative (6). Other findings at the pulmonary level are recurrent bronchoobstructive symptoms and obstructive sleep apnea syndrome (8); the latter improves with the use of CPAP or mandibular traction. People affected with micrognathia may also present airway obstruction based on glossoptosis of the tongue (10).
Patients with BOPS have an inadequate innate and adaptive immune response and are more predisposed to upper respiratory tract infections, pneumonia and acute otitis media. (1) The literature also reports cases of recurrent urinary infections, urinary retention and increased risk of kidney stones (2). Somatic mutations related to the ASXL1 gene have been associated with acute myeloid leukemia and appear to affect approximately 7% of people with myelodysplastic syndrome; they also present in sideroblastic anemia and Wilms tumor (2,10). A systematic review demonstrated the relationship between a somatic mutation of the ASXL1 gene and the risk of developing hematological cancers, including chronic myelomonocytic leukemia (up to 43% of cases), myelodysplastic syndrome (20%), myelo-proliferative neoplasms (10%) and acute myeloid leukemia (20%) (11).
If a diagnosis of BOPS is suspected, a sequencing of exons 12 and 13 of the ASXL1 gene, must be performed. (5). In the absence of a mutation identified in these exons of the ASXL1 gene, a somatic mosaicism or genetic heterogeneity is possible (other causal genes not identified to date) (2,12 ).
The BOPS prognosis is poor, with a high incidence of childhood mortality (2). Respiratory infections and recurrent wheezing episodes are common and represent 42% of deaths in the first two years of life (10). Death due to cardiovascular causes is associated with bradycardia and apnea, which represent four (33%) of the 12 deaths published in the literature (although none of these individuals had a molecular confirmation of BOPS) (2).
The treatment is symptomatic and focuses on clinical manifestations, cyclic vomiting can be controlled by identifying and avoiding the triggering factors (2). An earlyan enteral route such a gastrostomy has been proposed to reduce bronchoaspiration, improve nutrition and avoid unnecessary hospitalizations (10). Tracheostomy is recommended for patients with recurrent bronchoaspiration who develop secondary lung disease and those with severe sleep apnea who do not improve with non-invasive treatment (CPAP, BiPAP) or with surgery (for example, mandibular distraction) (2).

| summarizes the most |
| representative clinical characteristics. |
| Central Nervous | Ventriculomegaly, delayed myelination, delayed neurodevelopment, severe or profound |
| System | intellectual disability, Dandy-Walker malformation and seizures. |
| Osteomuscular | congenital dislocations. |
| Gastrointestinal | Functional constipation, chronic emesis and gastroesophageal reflux. |
| Cardiovascular | Defects of the atrial septum, persistent ductus arteriosus, valvular anomalies and pulmonary stenosis. |
| Respiratory | Repeated broncho-obstructive episodes and obstructive sleep apnea |
| Hematological | Acute myeloid leukemia, sideroblastic anemia, Wilms tumor and myelodysplastic syndromes. |
| Inmunological | Upper respiratory tract repeated infections |
| Typical Posture | Elbow, wrists and flexed metacarpophalangeal joints, ulnar deviation, of the hands and hypertonic limbs with central hypotonia. |