1.
Sturge-Weber Syndrome in a 28-Year-old Tanzanian Female: A Case Report Sarah Shali Matuja ? , Maryam Amour ? , Evangelista Malindisa ? & William Matuja ? Abstract-Background: Sturge Weber Syndrome is a sporadic neurocutaneous disorder of elusive etiology which is characterized by a vast continuum of manifestations ranging from neurological, cutaneous and ocular features. The main complications of the disorder include epileptic seizures, hemi paresis, and delayed neuropsychological development leading to poor quality of life.
Case presentation: We present a 28 years old female of African descent who was seen at our neurology outpatient clinic in Dar es Salaam, Tanzania with a chief complaint of relapsing generalized motor seizures for the past 1 month. She had been on Anti-Epileptic Drugs since birth with poor control. Physical examination revealed an obese lady with Port Wine Stain appearance on the left half of the head, face, and neck. She had reduced visual acuity on the left eye, dysphasia with severe right-sided spastic hemi paresis. Her Computed Tomography films revealed extensive gyral and sub cortical calcifications seen on the left posterior cerebral parenchymal also involving the parietal-temporal lobes. Her electroencephalography showed abnormal focal activity in keeping with a diagnosis of Sturge weber syndrome. The patient is currently on carbamazepine and clobazam with remarkable improvement attending regular follow-ups at our outpatient clinic.
Conclusions: Sturge weber syndrome is a rare congenital disorder presenting with a diverse spectrum of clinical manifestations. We present a case of delayed diagnosis thus recommending complete understanding of the disorder to clinicians in order to ensure early diagnosis since it offers opportunities to control for seizure-related complications leading to preserved quality of life.
Keywords: sturge weber syndrome, seizures, port-wine stain, angiomas.
2. I.
Background turge weber syndrome (SWS) is a sporadic neurocutaneous disorder of elusive etiology (1). It is characterized by a vast continuum of manifestations ranging from neurological, cutaneous and ocular features (2). In 1879, Sturge linked the dermatological and ophthalmic manifestations of the disorder to the neurological symptoms and signs, later in 1929 Weber described the radiographic changes associated with SWS (3). The estimated frequency is 1:50,000 live births yet experts believe more have the disorder, it is just undiagnosed (4). It has no racial predilection and has an equal male to female ratio (5). A proposed mechanism associated with SWS is a somatic mosaic mutation in the Guanine Nucleotide G Protein Q polypeptide gene (GNAQ) located at chromosome 9 which results in activation of several signaling cascades responsible for the syndromic manifestations of the disease (6,7).
Neurological malformations are characterized by presence of an ipsilateral leptomeningeal angioma responsible for the epileptic seizures, migraine like vascular headaches, hemiparesis, and mental retardation (8).
Facial Port wine stain (PWS), a dermo-capillary vascular malformation is the main cutaneous manifestation usually present at birth and its distribution involves the forehead and upper eyelid in the first (V1), second (V2) division as well as the third (V3) division of the trigeminal nerve (9,10). Ocular features include glaucoma or choroidal hemangiomas (11,12). They may also present with hypothalamic-pituitary disorders like growth hormone deficiency and central hypothyroidism (13,14).
Neuro imaging with either Magnetic Resonance Image or Computed Tomography scan (CT) is preferred to characterize the cerebral abnormalities which become apparent later in life (15). Treatment of SWS is usually conservative encompassing a combination of anti-epileptic drugs (AED) to ensure seizure control and management of glaucoma (16). In this report, we present a case of documented SWS in Tanzania with its classical clinical manifestations and neurological complications.
3. II.
4. Case Presentation
A 28 years old female of African descent was seen for the first time at our neurology outpatient clinic in Dar es Salaam, Tanzania with a chief complaint of relapsing generalized motor seizures on daily basis for the past 1 month. She was known to suffer from epileptic-seizures and had been on Anti-Epileptic Drugs (AED) of unknown dosage since birth (phenobarbital and valproate) with irregular clinic visits due to challenges in distance from home to the health care facility. Her seizures were not well controlled which ultimately led to cognitive impairment, delayed motor milestone development which hindered her from attending school. She was the 4 th born of 6 children and the other siblings were of good health.
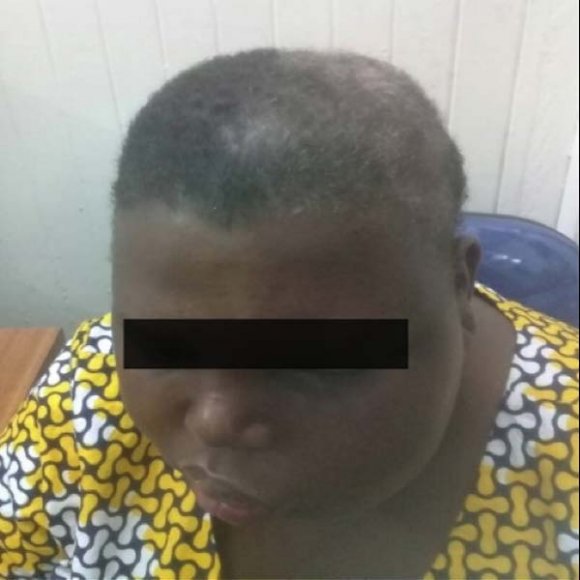
On examination, she was obese with a hyper pigmented lesion extending from the left half of the head, face, and neck consistent with a PWS which was present since birth (figures 1 and 2). She had reduced visual acuity on the left eye (N 26 ) probably due to glaucoma which was previously diagnosed during childhood and was treated medically. She had a poor understanding of spoken speech and was unable to communicate properly with severe right-sided spastic hemiparesis.
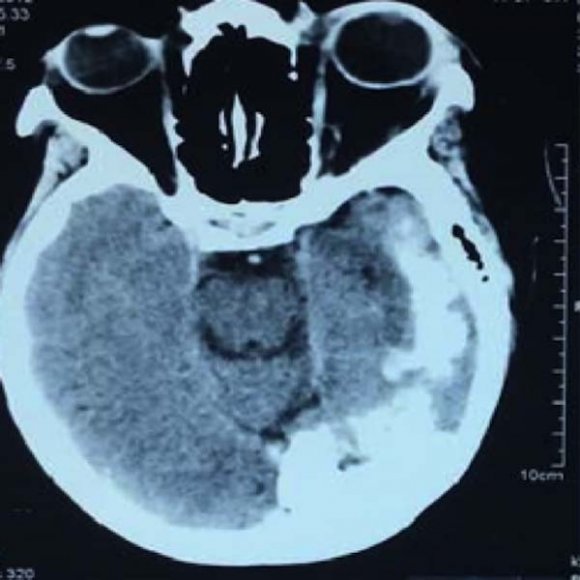
Her most recent brain CT films taken as an adult revealed extensive gyral and subcortical calcifications seen on the left posterior cerebral parenchymal extending to the left parietal and temporal regions (figures 3 and 4). Her electroencephalogram revealed an abnormal focal activity with normal thyroid and growth hormone assays. A diagnosis of SWS was made based on clinical and neuroimaging features. Phenobarbital and valproate were gradually stopped and the patient was subsequently commenced on carbamazepine 400mg twice daily (this was the maximum dose she could tolerate) and folic acid 5mg once a day. After 6 months of unsatisfactory response to monotherapy, clobazam 10mg twice daily was added. She has been seizure-free for 2 years and is followed up every 4 to 5 months at our neurology clinic in Dar es Salaam, Tanzania.
5. III.
6. Discussion
SWS is a rare congenital neurocutaneous disorder. It is classified into three sub-types based on areas that are affected by residual blood vessels using the Roach scale (17). Type 1, which is the complete form includes the presence of both facial and leptomeningeal angiomas and patients may have glaucoma; type 2 is characterized by presence of facial angiomas alone and may have glaucoma and type 3 includes presence of an isolated leptomeningeal angioma; usually with no glaucoma (17). Based on the Roach criteria, our patient was the classical type 1 case.
Leptomeningeal vascular angiomas occur in 10 to 20% of cases, they are usually unilateral affecting the pia-arachnoid membrane and any region of the cerebral hemispheres (8). The parietal and occipital areas are most commonly affected leading to abnormal blood flow patterns causing cortical irritation as a result of ischemia, hypoxia, and gliosis (18). This leads to epileptic seizures and early onset seizures by the age of 2 has been associated with a poor prognosis leading to impaired cognition and mental retardation (19,20). Our patient had cerebral vascular angiomas involving the left temporal, parietal and occipital regions as evidenced by the CT brain films. Furthermore, she developed earlyonset seizures which were poorly controlled and neither was she on regular clinic visits which led to progressive neurological dysfunction including hemiparesis, speech and learning disabilities.
The presence of PWS seen in about 70% of patients typically involves the ophthalmic division of the trigeminal nerve ipsilateral to the cerebral angiomatosis (9). The lesion presents at birth and its appearance and size changes with age (16). Our patient presented with PWS observed since birth present on the ipsilateral side of the angiomatosis extending from the face, neck and left arm. Likewise, some patients parallel to our case may present with ocular abnormalities such as glaucoma as an ophthalmic complication presenting in 40% of patients from early childhood (20).
Treatment of SWS is variable and it entails a multi-disciplinary approach based on its diverse clinical picture. The mainstay in preventing progressive neurological damage is through early detection and vigorous control of seizures, leading to preserved quality of life (21).
The ideal first line AED that is commonly used in SWS is carbamazepine or oxcarbazepine monotherapy and an additional second-line therapy may be considered in resistant cases before opting for surgical interventions (20,22). Seizures in our patient cannot be described as resistant since there was a lack of proper monitoring of her initial regimen which she commenced since birth. She was not on regular clinics; her dosing regimen was not clear nor did she have any documented side effects. Remarkably however is that there was a significant improvement after the addition of clobazam to carbamazepine and the patient is currently seizure-free with regular clinic visits.
7. IV.
8. Conclusions
SWS is a rare congenital developmental disorder presenting with a diverse spectrum of clinical manifestations. We present a case of delayed diagnosis, thus recommend a complete understanding of the disorder to health professionals to ensure early diagnosis and treatment since it offers opportunities to prevent and control for neurological deterioration. We also strongly emphasize on the use of a combination of AED with close monitoring of patients using carbamazepine and another second-line agent like clobazam to treat seizures in SWS which results in complete seizure remission.
Ethical approval: Ethical approval was sought from the Directorate of Family Care Medical Clinic.
Consent to publication: Written informed consent was obtained from the patient and parents for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Availability of data and materials: Not applicable.
Competing interests: The authors declare that they have no competing interests. Funding: Not applicable. Authors' contributions: SSM and WM took history, performed the physical examination, diagnosed and managed the patient. SSM wrote the initial draft of the manuscript. SSM, MM and EM critically reviewed the manuscript. All authors read and approved the final manuscript.