1. Introduction
hrombotic thrombocytopenic purpura (TTP/ Moscowitz Syndrome ) is a rare disorder leading to formation of microscopic thrombi throughout different systems of body due to endothelial injury causing platelet activation and coagulation cascade.
Majority of cases of TTP arise from ADAMTS13(an enzyme responsible for breaking down large multimers of vWFmultimers into smaller products) deficiency or inhibition. If it doesn't happen ultimately increase endothelial injury and platelet activation takes place resulting in widespread microtrombithrougout circulation. If not treated appropriately, thrombotic thrombocytopenic purpura-hemolytic uremic syndrome (TTP-HUS) in adults typically follows a progressive course resulting in irreversible renal failure, progressive neurologic deterioration, cardiac ischemia, and ultimately death of patient. Before the introduction of plasma exchange therapy it had a mortality of 90% which has been dropped to less than 10% in initial 6 months. Idiopathic thrombotic thrombocytopenic purpura is still a challenge to manage especially if it is unresponsive to plasma exchange therapy despite the evolution of various therapeutic options. [15,16].
A very rare form of TTP called Up Shaw Schulaman Syndrome can occur in some patients in which there is congenital defect in ADAMTS 13 but its presentation is usually very mild and manifests mostly in stressful circumstances.
2. II.
3. Pathogenesis
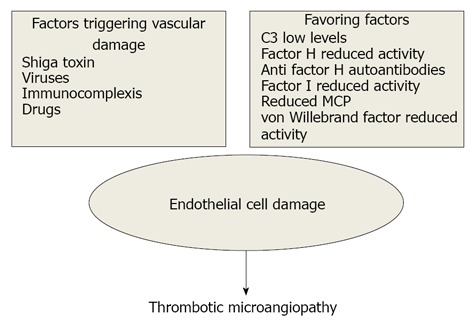
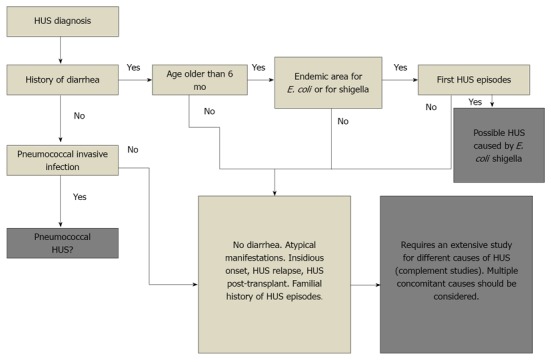
After an E. coli infection with production of Shiga-toxin, several factors influence the progression of Author ?: e-mail: [email protected] the disease to HUS. (1) Bacterial strain. For serotype O157/H7, the progression is approximately 15%; (2) Age: In children younger than 5 years, the rate of progression is 12.9%, while it is 6.8% in children between 5 and 10-year-old and 8% in children over 10 years; (3) Antibiotic therapy. Antibiotic therapy for E. coli O157/H7 infection may increase the risk of HUS, though a recent meta-analysis does not confirm this association; and (4) Environmental and genetic factors. Such factors are not yet fully understood, but several authors have suggested that factor-H abnormalities could influence the disease evolution in HUS D+. Shiga toxin is produced by E. coli in the gut and is then absorbed.
Shiga toxin consists of two subunits: A and B. Subunit A, approximately 32 kDa, is cleaved by proteolysis into two peptides: A1 (28 kDa) and A2 (4 kDa). In target organs (e.g., kidney, brain and gut), subunit B recognizes and binds to glycolipid receptors on the cell surface. In humans this receptor is Gb3 (globotriosilceramyde) and is highly expressed on kidney tubular cells, and brain and gut endothelial cells. Moreover, in the kidney cytotoxicity is further amplified by tumor necrosis factor-? (TNF-?).
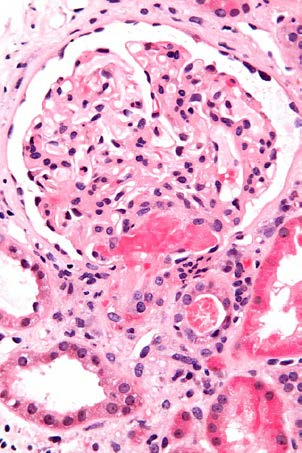
After binding to cell surfaces, Shiga toxin is endocytosed and retrograde-transported to the Golgi apparatus and the endoplasmic reticulum; it is then translocated to the cytosol where it inactivates ribosomes and causes cell death. [18] Micrograph showing an acute thrombotic microangiopathy, as may be seen in TTP. A thrombus is present in the hilum of the glomerulus (center of image). Kidney biopsy. Measurement of ADAMTS13 activity: The value of using information concerning ADAMTS13 for initial diagnosis and treatment decisions remains questionable, especially since such results may not be available for several days, well after initial treatment decisions need to be made. Also, ADAMTS13 activity measured by different methods (eg, using a synthetic substrate versus intact von Willebrand factor) may have discrepant results in some patients.
? It has been concluded that the group of patients with severe ADAMTS13 deficiency does not include all patients who may respond to plasma exchange, and that persistence of ADAMTS13 deficiency with or without inhibitory antibody activity following plasma exchange may still be consistent with an excellent clinical response to treatment. ? ADAMTS13 measurements appear to be of greatest clinical value for estimating the prognosis for relapse after recovery from an acute episode, for initial treatment of a patient who has an acute, recurrent episode, and perhaps also for helping to distinguish the different mechanisms of thrombotic microangiopathy in complex clinical situations. Patients with an ADAMTS13 activity >10 percent at the time of their initial presentation rarely relapse. However the clinical importance of ADAMTS13 measurements during remission is unknown. Approximately 20 percent of patients will have persistent ADAMTS13 deficiency that may not predict a risk for relapse.
ADAMTS13 activity can be low due to an inherited defect or an acquired inhibitor, such as an autoantibody. However, the absence of a demonstrable ADAMTS13 inhibitor does not exclude the diagnosis of acquired, autoimmune TTP-HUS. The titer of ADAMTS13 inhibitors may be too low to be observed in a plasma mixing study, or may have been neutralized by multiple transfusions prior to obtaining a sample. An acquired autoimmune etiology, rather than a congenital deficiency of ADAMTS13, is documented by recovery of ADAMTS13 activity to normal during remission or by documenting the presence of an inhibitor at the time of a relapse.
These observations support the current practice of requiring only thrombocytopenia and microangiopathic hemolytic anemia with no other apparent cause in order to make a clinical diagnosis of TTP-HUS and initiate prompt treatment with plasma exchange in appropriate scenarios
4. Use of Complement markers in differentiating typical and atypical HUS:
Testing of biomarkers of complement activation, specifically C5a and C5b-9, may be useful clinically to confirm the clinical diagnosis of aHUS and differentiate it from acquired TTP. Although limited by the retrospective nature of the data, they should form the basis for future studies to evaluate their role in the confirmation of the diagnosis as well as a tool to objectively monitor the response to complement inhibition therapy. [16] IV.
5. Discussion
The mainstay of treatment for most patients with TTP-HUS is plasma exchange, which in the context of this syndrome refers to the removal of the patient's plasma by pheresis and the replacement with donor plasma rather than another replacement fluid such as albumin. [7,8].Compared with the mortality rate of 90 percent prior to the use of plasma exchange, the mortality rate for patients treated with plasma exchange is 25 percent or less .Plasma exchange should be initiated even if there is some uncertainty about the diagnosis of TTP-HUS, as it is considered that the potential dangers of rapid deterioration from TTP-HUS exceed the significant risks of plasma exchange treatment. [1,2].Many patients with typical TTP-HUS respond very well to plasma exchange therapy and have a great prognosis but aHUSpatientsdon't respond very well to plasma exchange therapy in addition to immunosuppressant therapy. Eculizumab has currently modified the outcome of aHUS patients but it helps to reduce morbidity specially if it is used at an earlier time during the course of treatment. Several advances in the treatment of atypical hemolytic uremic syndrome (aHUS) have placed an increased emphasis on the rapid and accurate differentiation of aHUS from acquired thrombotic thrombocytopenic purpura (TTP).Although both disorders share the common end point of widespread microvascular disease and end organ injury, their distinct underlying mechanisms of microvascular injury likely explain their differing responses to plasma exchange (PEX) and the response of patients with aHUS to complement inhibition therapy. Despite advances in our understanding of both aHUS and acquired TTP in recent years, both diseases remain clinical diagnoses.
Plasma exchange therapy: Plasma exchange was found to be superior to plasma infusion in the treatment of the vast majority of adults with TTP-HUS in two landmark trials that included 210 patients. Plasma exchange is more effective than plasma infusion in most adults with "classical" TTP-HUS because for the great majority of those who have decreased ADAMTS13 activity, the decreased activity is due to an inhibitory antibody. Plasma exchange both removes the inhibitory antibody and supplies replacement ADAMTS13 from the donor plasma in UpshawSchulaman Syndrome patients. a) Eculizumab in refractory TTP patients Most patients with refractory or relapsing TTP receive additional immunotherapy but there are cases in which patients don't respond to plasmapharesis therapy and need other treatment modalities like eculizumab which has been proven effective in many cases refractory to combination of plasmapharesis and immunosppressive therapy.
Many patients with diarrheal/STEC-HUS have an especially severe course and are challenging to
6. ? Cost
? Plasmapheresis removes eculizumab from the circulation, which will impair its effectiveness. So when patient is shifted to eculizumab plasma exchange therapy has to be discontinued. [8,9].
V.
7. Concluion
Eculizumab use should be used for refractory TTP with normal ADAMTS 13 levels to plasma exchange therapy. Current guidelines suggest its use if patient fails to respond to plasma exchange after 2 weeks of daily but still not enough data is available to guide the duration of eculizumab use and use of complement testing to guide therapy. Early differentiation of typical versus atypical HUS can be very helpful in early administration and start of eculizumab therapy and can be very helpful for atypical HUS and effective in decreasing morbidity and ultimately progression to multisystem involvement like ESRD in many people.
8. b) Submission Declaration and Verification
We also declare that the work described has not been published previously except in the form of an abstract or as part of a published lecture or academic thesis or as an electronic preprint, is approved that, if accepted, it will not be published elsewhere in the same form, in English or in any other language, including electronically without the written consent of the copyright-holder.