1. Introduction
iabetic nephropathy (DN), a common and severe complication of Diabetes mellitus (DM), is the major cause of chronic kidney disease (CKD) which normally leads to end stage renal disease or dialysis. It is estimated that the number of people with diabetes will double by 2030 around the world, and the situation is more serious in developing country [1,2]. The mortality of dialysis patients with DN is higher than that of non-diabetic patient [3]. Thus, the thorough understanding of pathophysiology of DN will be one of the most important medical concerns in the future.
Numerous efforts have been made to investigate the molecular mechanism of DN with an aim to identify causative factors. The data indicated that hemodynamic and metabolic factors contribute to the development of DN [4][5][6]. Hemodynamic factors include alterations in flow and pressure, and the activation of renin-angiotensin system (RAS) [3]. Hyperglycemia related pathways are also activated, which lead to the formation of advanced glycation end products (AGEs), over-expression of protein kinase C (PKC), increased oxidative stress [5,6]. Clinical strategies based on some of these causative factors for preventing DN, include inhibition of RAS via angiotensin converting enzyme inhibitors (ACEI) or angiotensin receptor blockers (ARB); endothelin antagonists [7,8]. However, recent studies demonstrate that these clinical strategies only delay but cannot stop the progression of DN [9,10].
Advances in understanding of the pathogenesis and pathology of DN have made it clear that DN occurs as a result of imbalance between causative factors and endogenous protective factors (Fig. 1). Both aspects of DN mechanisms provide potential targets for disease prevention. To emphasize this concept, this review will focus on some of the current knowledge concerning both causative factors and endogenous protective factors.
2. Causative Factors
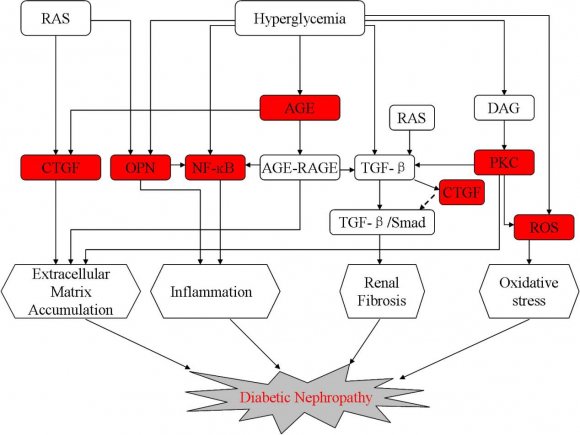
The most significant changes which characterize DN include glomerular and tubular hypertrophy, thickening of the peripheral glomerular basement membrane, mesangial expansion, glomerulosclerosis and tubulointerstitial fibrosis [11]. These structural changes occurs as a result of an interaction between hemodynamic and metabolic factors, and finally lead to increased glomerular filtration rate (GFR), proteinuria, systemic hypertension and the loss of renal function (4,12). Numerous efforts have been made to study the major causative molecules or pathways which include AGE, PKC, NF-?B, CTGF, ROS, Osteopontin (Fig. 2). Advanced glycation end products (AGEs) as a result of chronic hyperglycemia and oxidative stress have been postulated to play major roles not only in the development of DN, but also in a range of cardiovascular complications [13,14]. It is reported that AGE exert toxicity via three mechanisms: deposition, in situ glycation and receptor interaction [6]. Among these three mechanisms, the interactions between AGEs and their receptors (RAGE) play a major role in the progress of DM, especially DN. Its receptor is expressed on the surface of kidneys endothelial cells, podocytes, monocytes/macrophages, tubular and mesangial cells [15,16]. Binding of AGEs to the RAGE on these cell types will stimulate oxidative stress generation, activate intracellular molecules such as PKC, TGF -? , VEGF and NF-?B, evoke inflammatory and fibrogenic reactions, thereby causing progressive alteration in renal architecture and loss of renal function in DN [6,17]. The function of AGE-RAGE signaling pathway in the progress of DN has been proved by using the double transgenic mice mode which over expresses both iNOS and RAGE [18]. In this study, transgenic mice developed glomerular lesions rapidly, which could be prevented by AGEs inhibitor [18]. Soulis et al. (1996) initiated a research which also confirmed the beneficial effect of an AGE inhibitor Aminoguanidine in reducing the AGEs levels in blood and tissue of diabetic rats [19]. Similar beneficial effect was observed by using alagebrium, a putative AGEs cross-link breaker, to treat DM rodent model [20]. However, clinical trials for these AGE inhibitors were stopped due to toxicity of these inhibitors [21]. Thus, these studies provide further evidence that AGEs is a promising therapy target for DN and efforts should be made to find new inhibitor of AGEs for treatment of DN.
3. Volume XV Issue II Version
4. b) Protein Kinase c
Protein kinase C (PKC) belongs to the family of serine threonine kinase that act as an intracellular signal transduction system for many hormones and cytokines Diabetic Nephropathy: Causative and Protective Factors [22]. PKC has 15 different isoforms, many of which have been indicated to be involved in diabetic complications [9]. Among 15 isoforms of PKC, ?, ?, and ? isoforms have been most consistently implicated in DN. In DN, PKC isoforms, activated by enhanced diacylglycerol (DAG) and increased activity of polyol pathway, involves in numerous cellular pathways including NADH, ROS, Na+/K+ ATPase, Ang?, MAPK, VEGF, TGF-? and finally leads to series of physiological and structure changes such as endothelial dysfunction, glomerular basement membrane thickening, extracellular matrix accumulation, mesangial expansion, renal tubular fibrosis and glomerulosclerosis [9,10,23].
A range of novel compounds has been recently examined to inhibit PKC dependent pathways in DN. Ruboxistaurin, a selective inhibitor of the PKC-?, could normalize glomerular hyperfiltration, attenuate histological injury and functional decline, and reduce TGF-? levels and proteinuria [24]. A randomized clinical study has been carried out, in which the patients with DN took ruboxistaurin orally for one year. The study showed that DN patients treated with ruboxistaurin daily had a 24% greater decline in albuminuria than those given the placebo, and they had a stable estimated glomerular filtration rate as well [25]. In a recent study conducted by Bhattacharya et al. (2013), it was found that the upregulation and activation of PKC isoforms ?, ?, and ? in the renal tissue of diabetes rats play a detrimental role in the pathogenesis of DN by accumulating of extracellular matrix through upregulation of TGF-?, fibronectin and type ? collagen [23]. Treatment of diabetic rat with D-Saccharic acid 1, 4-lactone (DSL) could help to ameliorate alloxaninduced upregulation of PKC isoforms ?, ?, and ? as well as the accumulation of fibronectin and collagen [23]. Thus, strategies to target PKC pathway using isoform-specific inhibitors could be one of the promising therapeutic options, but well-designed large and longterm clinical studies are needed to establish its efficacy for prevention and treatment of DN.
5. c) Connective Tissue Growth Factor
Connective tissue growth factor (CTGF), known as insulin-like growth factor-binding protein 8 (IGFBP8) and CCN2, is increasingly being implicated in structural and functional changes of diabetic renopathy [26]. It is reported that the expression level of CTGF increased in glomerular and tubular of diabetes patients, and elevated in both early and late DN in humans [27]. CTGF, stimulated by both hyperglycemia related factors, such as AGEs, and hemodynamic stimuli such as angiotensin [28,29], is involved in mesangial cell hypertrophy, accumulation of extracellular matrix, epithelial-to-mesenchymal transition of tubular cells [27]. CTGF is also a fibrogenic cytokine in the kidney and it is known to be a downstream mediator of the profibrotic effects of TGF-? inducing renal fibrosis [30,31]. In TGF-? mediated renal fibrosis, the activated type 1 receptor of TGF-? phosphorylates and activates members of the receptor-Smads (R-Smads; Samd2 and Smad 3) which then form oligomers with the co-Smad and regulate the expression of target genes in nucleus; Smad7, an inhibitory Smad, prevents the recruitment and phosphorylation of Smad2 and Smad3 [12]. Several studies indicated that CTGF plays a central role in promoting the TGF-?/Smad signaling activity by decreasing the availability of smad7, which is inhibitory for Smad2 and 3 [27,32]. In an animal model of unilateral ureteral obstruction (UUO), it was found that CTGF antisense treatment could attenuate tubulointerstitial fibrosis which further confirms the role of CTGF on TGF-? inducing renal fibrosis [33]. In a study conducted by Adler et al (2010), it was found that FG-3019, a humanized anti-CTGF monoclonal antibody, could decrease albuminuria of diabetic patients with incipient nephropathy effectively [34]. These studies demonstrate that strategies specifically targeting CTGF to retard the development of renal disease are likely to be an excellent therapeutic strategy for DN, although prospective studies are lacking.
6. d) Nuclear Factor Kappa b
Nuclear factor Kappa B (NF-?B), a transcription factor, plays an important role in cell survival and its inhibition leads to apoptosis. In the latent state, NF-?B is sequestered in the cytosol by its inhibitor I?B [35]. Upon stimulations, its inhibitor I?B will be phosphorylated and degraded rapidly. Proteasomal degradation of I?B ultimately frees NF-?B which then translocates into nuclear and activates targeted gene [35]. Numerous studies indicated that NF-?B is important modulator of diabetic complications, especially in DN [36,37]. It is reported that NF-?B could be activated by a range of stimuli including high glucose, AGEs and ROS [38]. And activated NF-?B in turn regulates numerous genes including cytokines, adhesion molecules, NO synthase, angiotensinogen and other inflammatory factor implicated in the process of DN [39]. In addition, recent studies have indicated that NF-?B plays a key role in podocyte apoptosis [40], modulates the TGF-? intracellular signaling pathways [41], which provide further evidence for the role of NF-?B in the pathogenesis of DN. In a study conducted by Chiu et al. (2009), the typical characteristics of DN including mesangial expansion, accumulation of extracellular matrix were observed in rats injected with streptozotocin [42]. After treating these diabetic rats with curcumin, an inhibitor of NF-?B, these diabetes-associated abnormalities were ameliorated. Similar beneficial effects were observed by using Polydatin and Lycopene, the putative inhibitors of NF-?B signal pathway, to treat DN rats induced by streptozotocin [43,44]. However, approaches to inhibit NF-?B have not been explored fully in clinical studies, most likely due to the intimate
7. e) Osteopontin
Osteopontin (OPN), also known as secreted phosphoprotein 1, is a complex secreted glycoprotein that facilitates cell adhesion and migration by binding integrins with its RGD domain [45]. OPN has also been shown to play a prominent role in inflammation via promoting macrophage retention and activating macrophage [46]. Its role in DN has recently been examined in OPN gene knockout mice [47]. It was found that diabetic OPN null mice have decreased albuminuria, glomerular extracellular matrix, mesangial area and TGF-? compared with their respective diabetic OPN+/+ littermates [47], which indicates that OPN promotes diabetic renal injury in diabetic OPN+/+ mice. Besides, the upregulated expression of OPN in human and mice with diabetes has been observed [48,49]. And OPN, induced by hyperglycemia and lipopolysaccharides [49], is expressed in all glomerular cells including mesangial cells, podocytes, and endothelial cells [50,51]. These results suggest that OPN contributes to DN via damage the glomerular cells. Lorenzen et al. (2008) carried out an experiment to investigate the molecular mechanism of OPN on cultured podocytes [49]. They found that OPN could activate NF-?B pathway, increase the expression of urokinase plasminogen activator and matrix metalloprotease, and finally lead to increased podocyte motility. The similar study was conducted by Nicholas et al. (2010) in which the effect of OPN on cultured mouse mesangial cells was studied [47]. The result shows that OPN could promote the accumulation of glomerular extracellular matrix through upregulating TGF-?, ERK/MAPK and JNK/MAPK signaling. They also found that the expression of TGF-? induced by glucose was inhibited by OPN antibodies. Thus, OPN seems to be a critical contributor to the pathogenesis of DN. However, further studies will be needed to validate whether OPN is truly a causative factor for DN or not.
8. f) Reactive Oxygen Species
High reactive oxygen species (ROS), induced by hyperglycemia, plays a prominent role in the pathogenesis of diabetic complications, especially DN [52,53]. It is reported that ROS could be produced by various types of cells which include endothelial cells, mesangial cells, podocytes, tubular epithelial cells under hyperglycemic [1,54]. Produced ROS are capable of disturb physiological function of these cells both directly, by oxidizing and damaging cellular macromolecules such as DNA, protein lipid and carbohydrate, and indirectly through the stimulation of multiple pathways, such as PKC, polyol pathways, NF-?B, RAAS, and accumulation of AGEs [52,55]. Zhang et al. (2012) investigated the role of NADPH oxidase-derived ROS in cultured mesangial cell and found that high glucose could upregulate NADPH oxidase through JNK/NF-?B pathway and consequently produce ROS which finally contributes to glomerular mesangial cell proliferation and fibronectin expression [52]. They also use resveratrol, a polyphenolic phytoalexin, to treat high glucose induced mesangial cell and the results showed that resveratrol could inhibit mesangial cell expansion and fibronectin expression through blocking JNK/NF-?B/NADPH oxidase/ROS signaling pathways [52]. In another study, schizandrin, a blocker of NADPH oxidizeinduced ROS signaling, was utilized to treat murine mesangial cell cultured in high glucose media [56]. The result showed that schizandrin inhibits high glucose induced mesangial cell proliferation and ECM overexpression through attenuating ROS level. Furthermore, a large number of experimental studies have proved the beneficial effect of antioxidants, such as Vitamins C and E, superoxide dismutase, and catalase, in ameliorating DN [57]. However, it is also reported that ROS are involved in the regulation of renal hemodynamic and renal ion transport which is the key for maintaining basic function of kidney [58,59]. Therapeutic effect of ROS in preventing of DN is still debatable at this time.
9. III.
10. Endogenous Protective Factors
The role of endogenous protective factors in the development of DN has been investigated widely. In a clinical research conducted by Perkins et al. (2003), 368 type 1 diabetic patien ts with microalbuminuria were followed up for 12 years [60]. It was found that, among these diabetic patients, more than 60% of type 1 diabetic patients were free from significant diabetic complications which suggest the presence of endogenous protective factors. Meanwhile, these results indicate that endogenous protective factors protect the diabetic patients from the progression of DN via neutralizing effect of risk factors such as PKC, ROS, TGF -? etc.
11. a) Netrin-1
The netrin-1, a diffusible laminin-related secreted protein, is originally identified as a neuronal guidance cue which directs neurons and their axons to targets during the development of the nervous system [61]. Recent investigations indicate that netrin-1 is highly expressed in many tissues outside the nervous system, especially in vascular endothelial cells of kidney to attenuate inflammation [62]. An investigation conducted by Wang et al. (2008) showed that downregulation of netrin-1 correspond with the increased expression of MCP-1 and IL-6 and infiltration of leukocytes into the kidney [63]. Mice with partial netrin-1 deficiency experience more severe degree of ischemic kidney injury because of exacerbated inflammation [64]. Meanwhile, it is also reported that administration of recombinant netrin-1 in kidney could suppress inflammation and apoptosis in vivo [65].
DN is a manifestation of an ongoing chronic low-grade inflammation [66]. The role of netrin-1 in DN has been investigated recently and the result showed that over-expression of netrin-1 could protect transgenic mice during DN via attenuating inflammation [67]. In a study conducted by Tak et al. (2013), partial netrin-1 deficiency mice mode (Ntrn 1+/-) was introduced to investigate the role of netrin-1 protein in STZ induced diabetic mice [68]. The result showed that Ntrn 1+/mice revealed a more severe degree of DN compared with wild-type mice [68]. In addition, they found that treatment of DN with netrin-1 was associated with attenuated albuminuria and improved histological scores for DN. However, as most of these studies were done in animal model, further studies in clinic would be important to investigate its therapeutic function.
12. b) Adiponectin
Adiponectin, known as ACRP30 and GBP28, is an adipokine produced by white adipocytes and encoded by the APM1 gene in humans and rodents [69]. It has two receptors, AD1POR1 and ADIPOR2 which have been found to be widely expressed in liver, kidney, and endothelial cells [70]. Through interacting with its receptors AD1POR1 and ADIPOR2, adiponectin could mediate increased 5'adenosine monophosphateactivated protein kinase (AMPK) and activate peroxisome proliferator-activated receptor alpha (PPAR?), respectively [70]. Recently investigation indicated that adiponectin have insulin-sensitizing effects which include stimulation of fatty acid oxidation and glucose uptake in skeletal muscle and suppression of glucose production in the liver via activating of AMPK in the peripheral tissue [71,72]. They found that administration of adiponectin could lower circulating glucose levels without stimulating insulin secretion in both healthy and diabetic mice [72].
Besides, it is reported that adiponectin has a renoprotective effect in chronic renal disease including DN [73,74]. In an experiment conducted by Ohahsi et al. (2007), the result showed that urine albumin excretion, glomerular hypertrophy and tubulointerstitial fibrosis were significantly worse in adiponectin knockout mice compared to wild type after performing subtotal (5/6) nephrectomy [74]. Further study demonstrated that adiponectin knockout mice developed podocyte foot process effacement which is a key process involved in the initial development of albuminuria [75]. Sharma et al. (2008) also reported that administration of adiponectin to knockout mice could help normalize albuminuria and restore podocytes foot process effacement via activating of AMPK in podocytes [75].
These finding strongly supports the importance of adiponectin as a renoprotective factor. However, it is still unclear whether adiponectin will provide significant effects toward human DN.
13. c) Activated Protein c
Protein C, known as an anticoagulant factor, is activated by binding of thrombin to its receptor, thrombomodulin. After activation, it is reported that protein C confers cytoprotective effect in various disease models, including DN [76,78]. In diabetic patients and diabetic mice model, the function of endothelial thrombomodulin protein C system, which is in charge of activating protein C, is impaired and the level of activated protein C is reduced correspondingly [76,77]. The study conducted by Isermann et al., (2007) also reported that the reduction of activated protein C in diabetic mice is responsible for the initiation of DN and maintaining high activated protein C level could protect glomerular filtration barrier by preventing glucoseinduced apoptosis in endothelial cells and podocytes [76]. Besides, it is also reported that activated protein C have anti-inflammatory and fibrinolytic effects [79,80]. In unilaterally nephrectomized C57/B16 diabetic mice model, the urine total protein to creatinine ratio, proteinurine and renal fibrosis were ameliorated by administration of exogenous activated protein C [80]. They also indicated that the concentration of causative factors such as monocyte chemoattractant protein-1 (MCP-1), TGF-?1 and CTGF were decreased significantly in APC-treated mice compared with untreated mice [80]. Thus, APC appears to be a protective factor with anti-apoptosis, anti-inflammatory and fibrinolytic effects for DN and clinical studies are needed to validate its therapeutic role.
14. d) Insulin
Insulin is an important vasotropic factor which regulates the function of vascular cells, such as endothelial cells, macrophages, and podocytes, via binding to its receptors on these cells [10]. After binding to its receptors, insulin can activate the pathway of insulin receptor substrate (IRS)/PI3K/Akt/endothelial NO synthase (eNOS) and stimulate the production of NO which results in vasodilatation and anti-thrombosis in the short term, and can inhibit smooth muscle cell growth and migration chronically [81,82]. It is also reported that insulin could increases the expression of VEGF in several cell types, which in turn act as survival factor of podocytes, endothelial cells, and mesangial cells [83]. Furthermore, the studies indicated that insulin could prevent apoptosis through inhibition of transcription factor FoxO [84] developed albuminuria, effacement of podocytes foot processes, increased deposition of components of the basal membrane, and a higher frequency of programmed podocytes apoptosis compared to control animals [88]. The pathology was quite similar to that seen in DN. Thus, this finding strongly supports the importance of insulin signaling as a renoprotective factor and improving insulin sensitivity in glomerular tissue may decrease the risk for DN.
15. IV. Conclusions

![Figure 2 : Possible factors involved in induction and progression of diabetic nephropathy. RAS, rennin angiotensin system; AGE, advanced glycation end products; DAG, diacylglycerol; PKC, protein kinase C; CTGF, connective tissue growth factor; NF-?B, nuclear factor Kappa B; OPN, osteopontin; ROS, reactive oxygen species; TGF-?, transformed growth factor ? a) Advanced Glycation end ProductsAdvanced glycation end products (AGEs) as a result of chronic hyperglycemia and oxidative stress have been postulated to play major roles not only in the development of DN, but also in a range of cardiovascular complications[13,14]. It is reported that AGE exert toxicity via three mechanisms: deposition, in situ glycation and receptor interaction[6]. Among these three mechanisms, the interactions between AGEs and their receptors (RAGE) play a major role in the progress of DM, especially DN. Its receptor is expressed on the surface of kidneys endothelial cells, podocytes, monocytes/macrophages, tubular and mesangial cells[15,16]. Binding of AGEs to the RAGE on these cell types will stimulate oxidative stress generation, activate intracellular molecules such as PKC, TGF -? , VEGF and NF-?B, evoke inflammatory and fibrogenic reactions, thereby causing progressive alteration in renal architecture and loss of renal function in DN[6,17]. The function of AGE-RAGE signaling pathway in the](https://medicalresearchjournal.org/index.php/GJMR/article/download/901/version/100513/3-Diabetic-Nephropathy_html/8607/image-3.png)